English
EnglishDepolariseringen og den påfølgende muskelkontraksjonen i hjertet initieres, som i andre muskeltyper, ved at det genereres en elektrisk impuls. I hjertet genereres denne av pacemakerceller og i skjelettmuskler av den nevromuskulære endeplaten. Men depolariseringen i hjertet er til sammenligning ganske langvarig, rundt 200 millisekunder, i motsetning til skjelettmuskelfibrene der depolariseringen varer 1-2 millisekund.
Depolariseringen av pacemakercellene depolariserer nærliggende muskelceller. Under depolariseringen i muskelcellene, åpnes Na+-kanaler og membranpotensialet stiger fra –90mV (millivolt) til +30mV. Depolariseringen sprer seg så inn i T-tubuli. Konsekvensen blir at Ca2+ (kalsiumioner) frisettes fra sarkoplasmatisk retikulum og blir tilgjengelig for actinfilamentene. Så langt er reguleringen av muskelcellenes kontraksjon identisk med skjelettmuskelfibrenes.
Samtidig med at Na+-kanalene åpner under depolarisering i hjertemusklene, åpnes også langsomme Ca2+-kanaler slik at Ca2+ fra det ekstracellulære rom diffunderer langsomt inn i muskelcellen. Dette Ca2+ trigger ytterligere frisetting av Ca2+ fra sarkoplasmatisk retikulum. Som kjent lukker Na+-kanalene seg kjapt etter depolarisering, men i hjertets muskelceller forsetter depolariseringen selv om Na+-kanalene har lukket seg. Det er fordi Ca2+ sakte, men sikkert diffunderer inn (på grunn av langsomme kanaler). Vedvarende diffusjon av Ca2+ inn i muskelcellen skaper et platå i aksjonspotensialet, og så lenge Ca2+ diffunderer inn, vedvarer kontraksjonen. Samtidig med at Ca2+ diffunderer inn, er det minimal diffusjon av K+ (kalium) ut av muskelcellen. Summen av de to prosessene rundt Ca2+ og K+, er forklaringen på at man får en vedvarende depolarisering i hjertemuskelen – platået. Når kalsiumkanalene lukkes og kaliumkanalene endelig åpnes, får vi repolarisering.
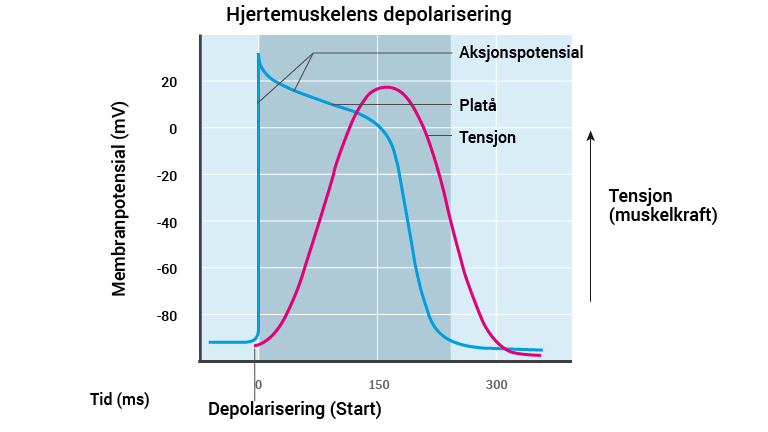
Fig.11
Merk av figur 11 at kraften i muskelcellene øker under platået. Det forklarer vi med vedvarende depolarisering og øket mengde Ca2+ i muskelcellene. Myofibrillene (actin og myosin) får tid og nok Ca2+ til å danne mange bindinger. Dette gir kraft.
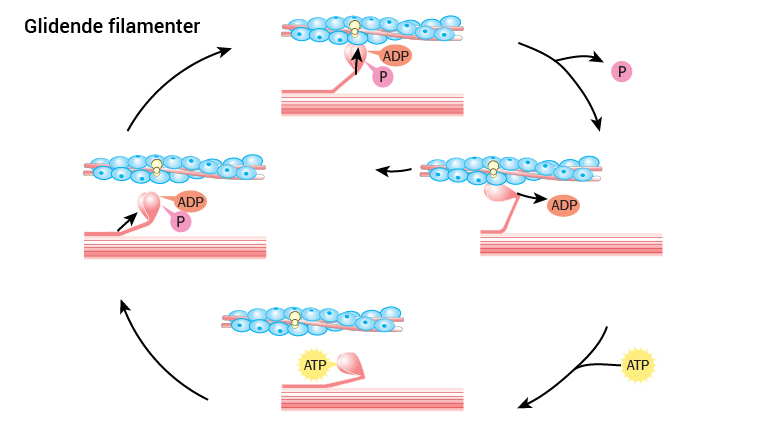
Selve muskelkontraksjonen, der myosin klatrer på actin, er identisk som kjent fra teamet om skjelettmusklene. Det følgende er derfor en kortfattet gjennomgang av denne prosessen:
Så lenge kalsium ikke er tilgjengelig for actinfilamentene i myofibrillene, kan ikke hodene på myosin binde actin. Men når Ca2+ blir frisatt vil det binde actinfilamentene på en slik måte at festepunktet for myosinhodene på actinfilamentene blir tilgjengelige. Dermed vil myosin feste seg til actin.
Hoderegionen på myosin har forut for bindingen på actin, bundet energimolekylet ATP og spaltet det til ADP og en fosfatgruppe. Det betyr at forut for at myosinhodet kan feste seg til actinfilamentene har ATP blitt spaltet. Det betyr at energi er blitt brukt (når ATP forbrukes, skjer det ved at en fosfatgruppe på molekylet spaltes av, se tema i cellebiologi for nærmere beskrivelse). Men både ADP og fosfatgruppen henger fortsatt på myosinhodet når den festes til actin. Når myosinhodet binder actin slipper myosinhodet fosfatgruppen. Da oppstår det er forandring i myosinets tredimensjonale struktur på en slik måte at hodet bøyes, og drar med seg eller forflytter actinfilamentene i lengderetningen. ADP forsvinner. Når dette er gjort, vil et nytt ATP binde myosinhodet, og først da slipper myosinhodet taket på actin. ATP spaltes til ADP og fosfatgruppe og myosinhodet oppnår sin opprinnelige tredimensjonale struktur. Vi er tilbake til utgangspunktet der myosinhodet binder actin ved tilstedeværelse av kalsium. På ny vil myosinhodet forflytte actinfilamentene ved å nærmest klatre på dem, og ved repeterte sykluser av denne prosessen vil actin-og myosinfilamenter gli over hverandre og forkorte lengden på sarkomeren.